
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


Ziprasidone
Ziprasidone (marketed as Geodon, Zeldox by Pfizer) was the fifth atypical antipsychotic to gain approval (February 2001) in the United States. It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia, and acute mania and mixed states associated withbipolar disorder. Its intramuscular injection form is approved for acute agitation in schizophrenic patients for whom treatment with just ziprasidone is appropriate.
Ziprasidone is also used off-label for depression, bipolar maintenance, mood disorders, anxiety, aggression, dementia, attention deficit hyperactivity disorder, obsessive compulsive disorder, autism, and post-traumatic stress disorder.

Ziprasidone synthesis: John A. Lowe, Arthur A. Nagel. Pfizer Inc. U.S. Patent 4,831,031 (1989).
The oral form of ziprasidone is the hydrochloride salt, ziprasidone hydrochloride. The intramuscular form, on the other hand, is the mesylate salt, ziprasidone mesylate trihydrate, and is provided as a lyophilized powder.
Ziprasidone, chemically named (5-[2-{4-(l,2-benzisothiazol-3-yl)piperizin- 0 1 -yl } ethyl] -6-chlorooxindole)hydrochloride hydrate, is a substituted benzisothiazolylpiperazine. The free base of ziprasidone has the following structure:

Ziprasidone and some of its uses are described by U.S. Patent Nos. 4,831,031 and 0 5,312,925.
Like clozapine and risperidone, ziprasidone is a highly potent and selective 5-HT2receptor and dopamine D2 receptor antagonist. Seeger, T.F. et al, J. Pharmacol. Exp. Ther.. 275(1): 101-1 13 (1995). Ziprasidone is characterized as an antipsychotic, but may also have anxiolytic and antidepressant effects due to its ability to inhibit serotonin and 5 noradrenaline reuptake. Davis, R. and Markham, A., CNS Drugs, 8(2):154-159 (1997). The therapeutic potential of ziprasidone may also be enhanced by its high affinity for the 5-HT1A, 5-HT1D, 5-HT2Creceptor subtypes. Seeger, T.F. et al, J. Pharmacol. Exp. Ther.. 275(1):101-113 (1995).
The metabolism of ziprasidone is complex. When administered orally to
30 healthy humans, the drug is extensively metabolized by at least four major pathways: 1) N- dealkylation of the ethyl side chain attached to the piperazinyl nitrogen; 2) oxidation at sulfur resulting in the formation of sulfoxide or sulfone; 3) reductive cleavage of the bensisothiazole moiety; and 4) hydration of the C=N bond and subsequent sulfur oxidation or N-dearylation of the benzisothiazole moiety. Prakash, C. et al, Drug Metab. Dispos.,
35 25(7):863-872 (1997). At least 12 human metabolites have been identified: ziprasidone sulfoxide (ZIP-SO); ziprasidone sulfone (ZIP-SO2); 3-(piperazine-l -yl)-l,2-benzisothiazole (BITP); BITP sulfoxide; BITP sulfone; 6-chloro-5-(2-piperazinJ-yl-ethyl)JJ-dihydro- indol-2-one; 6-chloro-5-(2- {4-[imino-(2-mercapto-phenyl)methyl]-piperazin- 1 -yl} ethyl)- 1 ,3-dihydro-indol-2-one; 6-chloro-5-(2- {4-[imino-(2-methylsulfanyl-phenyl)methyI]- piperazin-1-yl} ethyl)- l,3-dihydro-indol-2-one; S-methyl-dihydro-ziprasidone; S-methyl- dihydro-ziprasidone sulfoxide; dihydro-ziprasidone sulfoxide; and (6-chloro-2-oxo-2,3- dihydro-lH-indol-5-yl)acetic acid. Two metabolites, ZIP-SO and ZIP-SO2, both of which are formed by oxidation of the ziprasidone sulfur atom are discussed herein. These metabolites have the following structures:

Ziprasidone Sulfoxide (ZIP-SO)

Ziprasidone Sulfone (ZIP-SO2)
Both ZIP-SO and ZIP-SO2 are minor metabolites, and account for less than about 10% and less than about 3% of ziprasidone metabolites found in human urine, respectively. Prakash, C. et al, Drug Metab. Dispos.. 25(7):863-872 (1997). It has been reported that neither metabolite likely contributes to the antipsychotic activity of ziprasidone. Prakash, C. et al, Drug Metab. Dispos., 25(7):863-872 (1997). Indeed, it has been reported that ziprasidone metabolites in general are not active at the D2 and 5-HT2A receptor sites. Ereshefsky, L., JL Clin. Psvch.. 57(suppl. l l):12-25 (1996).
Ziprasidone offers a number of benefits, but unfortunately many adverse effects are associated with its administration. Examples of adverse affects of ziprasidone include, but are not limited to, nausea, somnolence, asthenia, dizziness, extra-pyramidal symptoms, akathisia, cardiovascular disturbances, male sexual dysfunction, and elevated serum liver enzyme levels. Davis, R. and Markham, A., CNS Drugs, 8(2): 154-159 (1997). These adverse effects can significantly limit the dose level, frequency, and duration of drug therapy. It is thus desirable to find a compound which possesses advantages of ziprasidone but fewer of its disadvantages.
3. SUMMARY OF THE INVENTION
This invention relates to novel methods using, and compositions comprising, ziprasidone metabolites, preferably, ziprasidone sulfoxide and ziprasidone sulfone. These metabolites, prior to the present invention, have been reported to have little or no in vivo activity. The present invention encompasses the in vivo use of these metabolites, and their incorporation into pharmaceutical compositions and single unit dosage forms useful in the treatment and prevention of disorders that are ameliorated by the inhibition of serotonin reuptake at 5-HT2receptors and/or the inhibition of dopamine reuptake at dopamine D2 receptors. Such disorders include psychotic and neuroleptic disorders. In a preferred embodiment, ziprasidone metabolites are used in the treatment or prevention of neuroleptic and related disorders in mammals, including humans.

-
Ziprasidone (5-(2-(4-(1,2-benzisothiazol-3-yl-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2-(1H)-indol-2-one) is a potent antipsychotic agent and is useful for treating various disorders including schizophrenia, anxiety and migraine pain. Ziprasidone has been approved by the FDA for treatment of schizophrenia and goes by the brand name Geodon™ in the United States. Ziprasidone has also been indicated as useful for treating Tourette’s Syndrome (United States Patent 6,127,373), glaucoma and ischemic retinopathy (EP 985414 A2), and psychiatric conditions including dementia of the Alzheimer’s type, bipolar disorders, mood disorders, panic disorders, agoraphobia, social phobia, panic disorder, post-traumatic stress disorder, acute stress disorder, substance-induced anxiety disorder, anxiety disorders not otherwise specified, dyskinesias and behavioral manifestations of mental retardation, conduct disorder, and autistic disorder (United States Patent 6,245,766).
-
United States Patent 4,831,031 describes a genus of compounds encompassing ziprasidone and the synthesis of such compounds. Another method for synthesizing ziprasidone is described in United States Patent 5,206,366. A method for specifically synthesizingziprasidone hydrochloride monohydrate is described in United States Patent 5,312,925. A method for synthesizing ziprasidone mesylate dihydrate is described in United States Patent 6,245,765; and a method for synthesizing ziprasidone mesylate trihydrate is described in United States Patent 6,110,918. United States Patents 5,338,846; 5,359,068; and 6,111,105 also describe methods for synthesizing ziprasidoneand/or intermediates therefore.
-
The structure of ziprasidone can be depicted as:

(H. Howard, et al., “Ziprasidone Hydrochloride”, Drugs of the Future1994, 19(6): 560-563. As can be seen from the structure above, the compound ziprasidone comprises a chlorine atom.
-
Methods of introducing halogens into organic compounds are summarized in many organic text books. For example, J. March,Advanced Organic Chemistry, 4th Edition, pp. 587-591, and references cited therein, has a discussion of halogenation chemistry. More specifically, formation of chloro-aromatic compounds are frequently formed by a variety of methods also well known to those skilled in the art, and again summarized in J. March, Advanced Organic Chemistry, 4th Edition, Chapter 11, “Aromatic Electrophilic Substitution”. The chemistry to add a halogen, or more specifically a chlorine, to an aromatic group is thus well known to those skilled in the art. It is also known that such chemistry usually results in some mixtures of molecules, one of which is commonly the unreacted starting material not containing the chlorine atom. Further, over-chlorination is a problem well known to those skilled in the art; it is common to form some dichloro-compound impurities when the mono-chloro is desired and some trichloro-compound impurities when the dichloro- is desired. Over-chlorination is typically controlled by limiting the amount of the chlorinating reagent used. Unfortunately, control of over-chlorinated analogs in the drug substance by limiting the amount of chlorinating reagent utilized in the introduction of the aromatic chlorine substituent would be expected to result in more of a des-chloro impurity (unreacted starting material not containing the chlorine atom).
-
-
6-chlorooxindole (6-chloro-1,3-dihydro-2H-indol-2-one).
-
Although there are many known routes to 6-chlorooxindole, starting materials therefore are typically a substituted 4-chlorotoluene or 1,4-dichloro-nitrobenzene (see, G. J. Quallich and P. M. Morrissey,Synthesis, 1993, 51-53; and references cited therein; and F. R. Busch and R. J. Shine, “Development of an Efficient Process to 6-Chlorooxindole”, presented at the 208th ACS National Meeting in Washington D.C. in the Symposium on Technical Achievements in Organic Chemistry, 1994, (talk #126).). However, the concept of controlling chlorinated isomers, over-chlorination, or des-chloro impurities for the synthesis of 6-chlorooxindole is not described in the prior art. Other methods of synthesizing 6-chlorooxindole can be determined by a person of ordinary skill in the art, and such methods are included in the step of obtaining a batch of 6-chlorooxindole for the above-described method of this invention. Furthermore, a batch of 6-chlorooxindole can be obtained by purchase from manufacturers of organic chemicals, for example Plaistow, Ltd., Little Island, County Cork, Ireland or Finorga, Route de Givors, 38670 Chasse-Sur-Rhone, France.
-
Ziprasidone has two major fragments, benzisothiazol and substituted oxindole. In from 2 – mercapto acid methyl ester ( 1 ), the alkaline conditions with hydroxylamine-O-sulfonic acid reaction ring closure under alkaline conditions to obtain 5 . 5 3 can also be prepared from the disulfide, disulfides 3 by three methods (anthranilic acid by diazotization pass sulfur dioxide gas, o-fluorinated thiol acid and two xenon reaction, or dibromoethoxychlorophosphonazo acid and sulfur in copper iodide reaction), 3 and chlorinated sulfoxide and sulfone chlorination reaction of 4 , 4and ammonia reaction again 5 . 5 by chlorination with phosphorus oxychloride, the reaction of piperazine 7 . 7 may be made of the compound 8 ( 8 can be from 2 – cyano bromobenzene After the i-PrMgCl, ZnBr 2 , S 2 Cl 2 prepared in one-pot reaction) was prepared in DMSO and directly in the hot reaction piperazine.
Oxindole fragment from 6 – chloro-indol-2 – one ( 10 ) starts, the FC acylation later reduction with triethylsilane 12 , 12 and 7 occurs in alkaline aqueous solution S N 2 reaction with hydrochloric acid salt to obtain ziprasidone hydrochloride.
United States Patent 5,206,366,



MORE INFO UPDATED
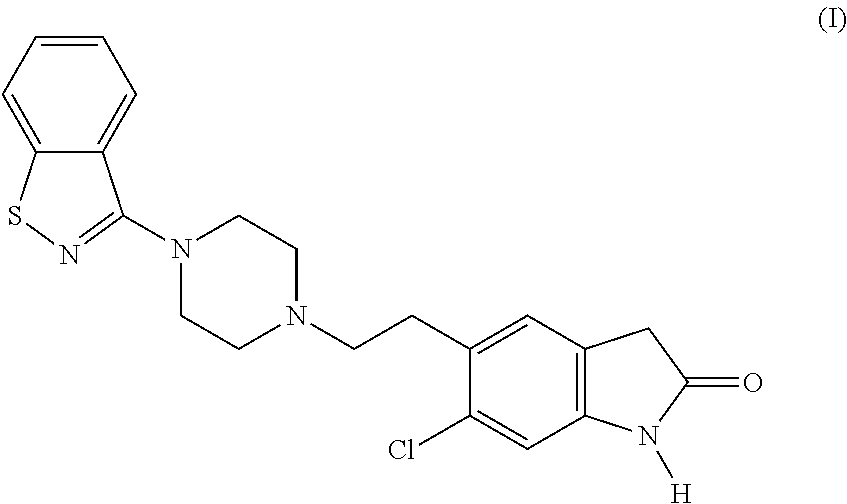
Ziprasidone is an antipsychotic agent with the following chemical name: 5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one of formula (I)
Ziprasidone is disclosed in U.S. Pat. Nos. 4,831,031 and 5,312,925 (assigned to Pfizer). Ziprasidone inhibits synaptic reuptake of serotonin and norepinephrine. No appreciable affinity was exhibited for other receptor/binding sites tested, including the cholinergic muscarinic receptor. The mechanism of action of ziprasidone, as with other drugs having efficacy in schizophrenia, is unknown. However, it has been proposed that this drug’s efficacy in schizophrenia is mediated through a combination of dopamine type 2 (D 2) and serotonin type 2 (5HT 2) antagonism.Ziprasidone’s antagonism of histamine H receptors may explain the somnolence observed with this drug.
U.S. Pat. No. 5,312,925 (Pfizer Inc.) describes a process for the synthesis of monohydrate of 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride and its characterization based on IR, XRD and moisture content. The ‘925 patent also discloses that the hemihydrate may be obtained by the process described in Example 16 of U.S. Pat. No. 4,831,031 and its characterization by IR, XRD and moisture content. It also discloses the IR, XRD and moisture content of anhydrous Ziprasidone hydrochloride. According to the invention in the ‘925 patent, Ziprasidone of water content of 3.97, 2.55 and 0.37% were used for the IR and XRD study of Ziprasidone hydrochloride monohydrate, hemihydrate and anhydrous. In this invention, the monohydrate ofZiprasidone hydrochloride was prepared by reacting anhydrous 5-(2-(4-(1,2-benzisothiazol-3-yl)piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one with aqueous hydrochloric acid. The temperature range of the reaction was maintained between 60 to 65° C. and aqueous hydrochloride used for salt formation was around 0.7 M. Depending on the reaction temperature and other conditions, the reaction times were set around 3 to 24 hours. The final product thus obtained was dried carefully in monitored conditions to make certain that water content was from about 3.8% to about 4.5% to obtain the stable monohydrate.
U.S. Pat. No. 6,150,366, discloses a manufacturing process of ziprasidonehydrochloride monohydrate, comprises: 1) dissolving, ziprasidone free base in a solvent comprising THF and water, in a volume ratio of about 22-35 unit volumes of THF to about 1.5-8 volumes of water; 2) heating the solution resulting from step (1); 3) adding HCl to the solution resulting from step (2); and 4) cooling the solution resulting from step (3) and crystals collected by filtration and drying.
U.S. Pat. No. 5,206,366 and U.S. Pat. No. 5,338,846 describe a process for preparing ziprasidone by reacting 1-(1,2-benzisothiazol-3-yl) piperazine with 5-(2-chloroethyl)-6-chloro-oxindole in water with a neutralizing agent such as sodium carbonate under reflux.
J. Med. Chem. 1996, 39, 143-148 discloses preparation of ziprasidone by reacting 1-(1,2-benzisothiazol-3-yl)piperazine with 5-(2-bromoethyl)-6-chloro-oxindole in isoamyl alcohol solvent in the presence of sodium carbonate.
Some salts of ziprasidone, and in particular, its hydrochloride salt is a potent commercial antipsychotic agent useful in the treatment of various disorders, including schizophrenia and anxiety diseases. Ziprasidone hydrochloride is currently marketed under the proprietary name of Geodon. Other salts ofziprasidone are also reported to be effective for the treatment of the same type of diseases.
Some of the processes described in the aforementioned patents necessitate the use of ion-exchange catalyst (i.e. sodium iodide) and/or phase transfer catalysts (for example tetra butyl ammonium bromide or tetra butyl phosphoriium bromide) in order for the coupling reaction producing ziprasidone to take place. For example, U.S. Pat. No. 4,831,031 indicates that arylpiperazinyl-ethyl (or butyl)-heterocydic compounds may be prepared by reacting piperazines of the formula II with compounds of the formula III as follows in [Scheme 1]:

Wherein Hal is fluoro, chloro, bromo or iodo; and Ar, n, X and Y are as defined therein with reference to formula I. According to the ‘031 patent the coupling reaction is generally conducted in a polar solvent, such as a lower alcohol, dimethylformamide or methylisobutylketone, and in the presence of a weak base and that, preferably, the reaction is carried out in the presence of a catalytic amount of sodium iodide, hydrogen chloride and neutralizing agent such as sodium carbonate.
In some instances, the ziprasidone obtained was purified by column chromatography, thus making the process impractical for large-scale preparations. Another process uses potentially explosive gases such as hydrogen in the presence of catalysts, for example zinc, palladium on carbon, followed by acid treatment to carry out a reduction and cyclization of an intermediate, in order to obtain ziprasidone.
Despite various processes disclosed in the prior art for the preparation of ziprasidone and salts thereof, still there is a need for a good process for producing ziprasidone and pharmaceutically acceptable acid addition salts of ziprasidone thereof, in high purity. One of the major problems faced in the prior art is formation of sticky material and difficult stirrability of the reaction mass. This problem is especially acute in large scale manufacturing.
picked up from polish site…translation is machine, please bear for errors
The first stages are two simple reactions: reduction of Wolf-Kiżnera and Friedel-Crafts

The next step is to reduce the use of triethylsilane and trifluoroacetic acid [2], and then the coupling with a compound 5 to obtain the final product.

a) method 1

b) method 2

In the second method, we have marked with an interesting transition.As you zoom scale but found that method 2 is the only possible one. Just destroy the product hydrochloride and get a clean API ready for tableting. [1] Bhugra D. The global prevalence of schizophrenia .. “PLoS Medicine”. 5 (2), pp. E151, 175 [2] Tetrahedron Letters “Selectivities in Ionic Reductions of Alcohols and Ketones with Triethyisilane / Trifluoroacetic Acid” 38, (6), 1997, pp. 1013-1016
1H NMR PREDICT

13C NMR PREDICT

http://www.google.com/patents/EP1476162A1?cl=en
Scheme 1
Stepl

MW = 167.59 MW = 244.08 Step 2

MW = 244.08 MW = 230.09 Step 3

MW = 230.09 MW = 255.76

MW = 412.94 Step 4

ziprasidone hydrochloride
MW = 412.94 monohydrate MW = 467.42
Scheme 2

Example 1 : Synthesis of Ziprasidone
Step 1 : Friedel-Crafts Acylation of 6-chloro-1 ,3-dihydro-2H-indol-2-one
Methylene chloride (310 L) and aluminum chloride (172.3 kg) were combined. Chloroacetyl chloride (66.7 kg) was added, and the resulting mixture was stirred for
45 minutes. 6-Chloro-1 ,3-dihydro-2/-/-indol-2-one (61.8 kg) was added. The reaction mixture was stirred at 28 to 32° C for 19.5 hours and then cooled to 15 to 20 C. Water (805 L) was cooled to 5 to 10 C. The reaction was quenched by the slow addition of the reaction mixture to the cold water. After the quench was complete, the mixture was heated to reflux, and the methylene chloride was removed by atmospheric distillation at 43 to 57° C. The resulting mixture was cooled to 15 to 20° C and stirred for 1 hour. The solids were isolated by filtration and washed with water (114 L) followed by methanol (114 L).- The solids were dried in a suitable dryer.
6-Chloro-5-(chloroacetyl)-1,3-dihydro-2/-/-indol-2-one, yield: 91.3 kg (101.4%). Note: A weight yield in excess of 100% resulted due to small amounts of residual salts which were removed in the following step.
The resulting 6-chloro-5-(chloroacetyl)-1,3-dihydro-2H-indol-2-one was carried through the following step in portions, one of which is detailed below.
Step 2: Trifluoroacetic Acid/Silane Reduction of 6-Chloro-5-(chloroacetyl)-1 ,3- dihydro-2H-indol-2-one
Trifluoroacetic acid (278 kg) and (74.2 kg) were combined and stirred slowly at 24 to 28° C. Triethylsilane (77.9 kg) was charged to the stirring mixture. The reaction temperature was allowed to exotherm slightly during this addition and was maintained between 50 to 62° C during the reaction period. , The reaction mixture was stirred for 8 hours, cooled to 38 C, and sampled for reaction completion. The reaction mixture was stirred at 50 to 54° C for an additional 3 hours. After the reaction was determined to be complete, the reaction mixture was cooled to 18° C, and quenched with water (594 L). The resulting slurry was stirred for 30 minutes at 10 to 15° C, and the solids were isolated by filtration. The product was rinsed from the tank and the product cake was washed with water (83 L) followed by methanol (76 L).
In each of two batches of equal size, tetrahydrofuran (742 L), Darco KB-B (1.9 kg), and the wet product cake were combined and heated to reflux. The resulting mixture was stirred at reflux for 30 minutes and filtered through a sparkler filter (pre- coated with filteraid) at 50 to 60° C to remove the carbon. The tank and sparkler were rinsed with hot tetrahydrofuran (38 L). Following the filtration the two batches were combined. The solution was concentrated in vacuo and stirred at 4 to 5° C for 1 hour. The solids were isolated by filtration and washed with cold tetrahydrofuran (38
L). The solids were dried in vacuo at 45 to 73° C until a loss on drying of 0.45% was achieved, giving 6-Chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one, yield: 60.1 kg (85.9%).
The resulting 6-chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one was combined with material of comparable quality and carried through the following step.
Step 3: Coupling of 6-Chloro-5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one and 3-(1-Piperazinyl)-1 ,2-benzisothiazole Monohydrochloride
Water (780 L) and sodium carbonate (126.0 kg) were combined and the mixture was stirred to dissolve. 3-(1-Piperazinyl)-1 ,2-benzisothiazole monohydrochloride (155.0 kg) and 6-chloro-5-(2-chloroethyl)-1 ,3-dihydro-2W-indol-2- one (150.4 kg) were added, and the reaction mixture was heated to reflux (-100° C). After 24 and 28 hours, the reaction slurry was sampled for reaction completion assay. The reaction was determined to be complete after the assay of the second sample. Water (1251 L) was added and the slurry was cooled to temperatures between 18 to
22° C. The solids were isolated by filtration and washed with water (302 L). The water wet solids were combined with isopropanol (940 L) and the resulting mixture was stirred for approximately 2 hours at ambient temperature. The solids were isolated by filtration, washed with isopropanol (89 L), and dried in vacuo at less than 43° C, giving 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3- dihydro-2H-indol-2-one, yield: 202.8 kg (80.8%).
The resulting 5-[2-[4-(2,3-benzisothiazol-3-yl)-1 -piperazinyl]ethyl]-6-chloro- 1 ,3-dihydro-2/- -indol-2-one was divided into two portions. These batches were carried separately through the following additional purification and resulted in material of comparable quality. The processing of one of these batches is detailed below.
Step 3R: Purification of 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]- 6-chloro-1 ,3-dihydro-2H-indol-2-one 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H- indol-2-one (51 kg), filteraid (4 kg) and tetrahydrofuran (2678 L) were combined. The mixture was heated to reflux (-65° C) for -1 hour, filtered while maintaining the temperature above 55° C, and rinsed with tetrahydrofuran (570 L). The product rich filtrate was partially concentrated in vacuo. 5-[2-[4-(2,3-Benzisothiazol-3-yl)-1- piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H-indol-2-one (51 kg), filteraid (4 kg) and tetrahydrofuran (2675 L) were combined. The mixture was heated to reflux (-65° C) for -1 hour, filtered while maintaining the temperature above 55° C, and rinsed with tetrahydrofuran (560 L). The product rich filtrate was combined with the partially concentrated mixture above and concentrated in vacuo. The resulting mixture was cooled to 0 to 5° C. The solids were isolated by filtration, washed with filtered tetrahydrofuran (113 L), and dried in vacuo at less than 41° C, giving ziprasidone, free base, yield: 79.3 kg (77.7 %).
A portion of the batch was combined with material of comparable quality which had been recrystallized separately and the batch was carried through the following step.
Example 2: Crystallization Salt Formation of Ziprasidone Hydrochloride Monohydrate Tetrahydrofuran (2715 L), water (307 L), and 5-[2-[4-(2,3-benzisothiazol-3-yl)-
1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2 – -indol-2-one (100.0 kg) were combined, heated to reflux (- 64° C), and stirred for -30 minutes. The solution was filtered and rinsed with tetrahydrofuran (358 L).
Water (203 L) and concentrated hydrochloric acid (29 L) were combined and stirred at ambient temperature. The resulting aqueous hydrochloric acid solution was charged to the 5-[2-[4-(2,3-benzisothiazpl-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3- dihydro-2 – -indol-2-one solution over a period of 27 minutes. The reaction mixture was cooled .to temperatures between 1 and 5° C over a period of -2 hours. The mixture was stirred between 1 and 5 C for -10 hours. The solids were isolated by filtration, washed with cold tetrahydrofuran (358 L), and dried until a water content of
4.1% was obtained.
Ziprasidone Hydrochloride Monohydrate, yield: 108.6 kg (96.0 % weight yiplrl)
The solids were milled on a Bauermeister mill. Example 3: Purification of 6-Chloro-5-(2-chloroethyl)-1,3-dihydro-2H-indol-2- one To Remove 5-(2-Chloroethyl)-1,3-dihydro-2H-indol-2-one
A 100 mL round bottom flask equipped with a magnet stirrer and reflux condenser was charged with 4.0 g (17.4 mmoles) of 6-chloro-5-(2-chloroethyl)-1 ,3- dihydro-2H-indol-2-one (Compound 3) and 36 mL of acetonitriie and 4.0 mL of water were added. The slurry was gently heated and stirred overnight (-18 hrs at -78° C). The heating was then removed and the slurry cooled to 0 to 5° C, and stirred for an additional hour. The product was collected by filtration, washed with a small portion of acetonitriie and the product dried under vacuum at 50° C, to give 3.77 g (94.3% yield) of 6-chloro-5-(2-chloroethyl)-1,3-dihydro-2H-ιndol-2-one. The level of the des- chloro impurity had been reduced from 1280 ppm to 230 ppm.
Example 4: Experimental Determination of Purge Factor for Compound 6 (1,3-Dihydro-2/Y-indol-2one)
A batch of 6-chloro-1 ,3-dihydro-2W-indol-2-one which contained a very high content of 1 ,3-dihydro-2λ7-indol-2-one was selected. This was intentionally selected so that higher levels of the impurity would be easier to measure, and to determine the purge factor for this impurity. An additional reason for this strategy of starting with material which was very high in the impurity for purposes of determining the purge factor of the impurity was to avoid having the material purge to less than the limit of analytical detection during the synthesis; thus resulting in a zero value in the final product. Since the purge factor is a ratio, it is not meaningful to divide by a zero result. (The material with the high level of impurity was used for this experiment but was NOT subsequently used in any studies with human subjects.) A batch of 6- chloro-1 ,3-dihydro-2H-indol-2-one which contained 4000 ppm of jl ,3-dihydro-2 – – ιndol-2-one was processed through the standard synthetic process according to Examples 1 and 2 above.
Following the first two steps of the synthesis, the level of the corresponding des-chloro impurity was measured, using the method described. It was found that
1700 ppm of 5-(2-chloroethyl)-1 ,3-dihydro-2H-indol-2-one (Compound 8 of Scheme 2, above) was present in 6-chloro-5-(2-chloroethyl)-1,3-dihydro-2 – -indol-2-one (Compound 3 of Scheme 1 , above). The processing was continued to 5-[2-[4-(1 ,2)- benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1 ,3-dihydro-2H-indol-2-one hydrochloride monohydrate, where it was determined that 600 ppm of 5-[2-[4-(1,2)- benzisothiazol-3-yl)-1-piperazinyl]ethyl]-1 ,3-dihydro-2H-indol-2-one (Compound 9 of Scheme 2, above) was present.
Thus the purge factor through the entire synthesis for the des-chloro analogs was from 4000 ppm to 600 ppm, or approximately a 6-fold decrease. Minor run to run variations in processing can lead to small differences in the yield and quality of the materials produced. A 20% error in the reproducibility of the impurity formation, that is if 500 ppm in one run expecting between 400 and 600 ppm in other experiments, is then allowed for. In the case of the synthesis described in Examples 1 and 2, with 5 processing steps, the additive experimental error could result in as much as a 2-fold difference in the level of the impurity. Thus, for the purpose of setting the upper limit, where the drug is going to be used by human subjects a conservative 3-fold purge factor was utilized. Therefore, to insure that the product produced would not contain over 100 ppm of 5-[2-[4-(1 ,2)-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-1,3-dihydro-2H- indol-2-one (Compound 9), a limit of 300 ppm of 1 ,3-dihydro-2H-indol-2-one
(Compound 6) in 6-chloro-1 ,3-dihydro-2H-indol-2-one (Compound 1) was determined.
…………………………………………..

The condensation of 1-(1,2-benzoisothiazol-3-yl)piperazine (I) with 6-chloro-5-(2-chloroethyl)-2-indolinone (II) in refluxing water or refluxing methyl isobutyl ketone gives the target indolinone derivative.
AU 8812537; EP 0281309; JP 1988301861

Wolff-Kishner reduction of 6-chloroisatin (I) gives 6-chlorooxindole (II), which is treated with chloroacetyl chloride under Friedel-Crafts conditions to yield 5-chloroacetyl-6-chlorooxindole (III). The ketone (III) is reduced using triethylsilane in trifluoroacetic acid to produce 6-chloro-5-(2-chloroethyl)oxindole (IV). 1,2-Benzisothiazolin-3-one (V) is converted to 3-chloro-1,2-benzisothiazole (VI) using phosphorus oxychloride and is then condensed with piperazine to provide 1-(1,2-benzisothiazol-3-yl)piperazine (VII). Finally, intermediate (VII) is alkylated by compound (IV) in the presence of sodium carbonate in water and is converted to the salt with aqueous hydrochloric acid.
US 4831031

WO 9500510
The nitration of 2,5-dichlorotoluene (I) with HNO3 in H2SO4/AcOH gives 2,5-dichloro-4-methylnitrobenzene (II), which is treated with t-butoxybis(dimethylamino)methane (III) in refluxing THF to yield 2,5-dichloro-4-[2-(dimethylamino)vinyl]nitrobenzene (IV). The condensation of (IV) with 1-(1,2-benzoisothiazol-3-yl)piperazine (V) in AcOH affords the disubstituted piperazine (VI), whose double bond is reduced by means of NaBH(OAc)3 in dichloroethane/AcOH to provide the saturated compound (VII). The condensation of (VII) with dimethyl malonate (VIII) by means of KOH in NMP gives the alkylated malonic ester (IX), which is hydrolyzed and monodecarboxylated with refluxing 3N HCl to yield the phenylacetic acid (X). The esterification of (X) with SOCl2 and methanol affords the methyl ester (XI), which is finally cyclized to the target indolone by reduction of its nitro group with sodium hydrosulfite in refluxing THF/ethanol. Alternatively, compound (VII) can be condensed with methyl cyanacetate (XII) by means of KOH in NMP to give the alkylated cyanacetic ester (XIII), which is hydrolyzed with refluxing 3N HCl to afford the already reported phenylacetic acid (X).
……………………
J Label Compd Radiopharm 1994,34(2),117
The Friedel Crafts condensation of 6-chloroindolin-2-one (I) with 14C labeled 2-chloroacetyl chloride (II) by means of AlCl3 in CS2 gives 6-chloro-5-(2-chloroacetyl)indolin-2-one (III), which is reduced with trimethylsilane in TFA to yield the labeled chloroethyl derivative (IV). Finally, this compound is condensed with 3-(1-piperazinyl)-1,2-benzoisothiazole (V) by means of Na2CO3 in refluxing water to provide the target radiolabeled compound.

………………….

The bromination of 3-chloro-1,2-benzoisothiazole (I) with Br2 in AcOH using FeCl3 as catalyst gives a mixture of 3,5-dibromo-1,2-benzoisothiazole (II) and 3,7-dibromo-1,2-benzoisothiazole (III) that are separated by flash chromatography. The desired isomer (III) is condensed with piperazine (IV) in refluxing diglyme to yield 7-bromo-3-(1-piperazinyl)-1,2-benzoisothiazole (V), which is condensed with 6-chloro-5-(2-chloroethyl)indolin-2-one (VI) by means of Na2CO3 in refluxing water to afford the brominated adduct (VII). Finally, this compound is debrominated with tritium gas over a Pd/BaSO4 catalyst in THF to provide the target radiolabeled compoun
……………….

The current process for ziprasidone involves preparation and isolation of the key intermediate 6-chloro-5-(2-chloroethyl)oxindole. An improved process for the synthesis of this intermediate is reported here. The new process involves use of a novel Lewis acid-mediated selective deoxygenation of the precursor ketone with tetramethyldisiloxane. The new method affords the desired compound in a one-pot process obviating the need for isolation of the potentially hazardous precursor ketone. This process was successfully scaled up to multikilo scale.

A new, one-step commercial process for the preparation of 3-(1-piperazinyl)-1,2-benzisothiazole, a key intermediate in the synthesis of ziprasidone has been developed: The reaction of 2-cyanophenyl disulfide (I) with piperazine (II) by means of DMSO and isopropanol at 120-5 C.
…………………………..

J Med Chem 1996,39(1),143
A new synthesis for ziprasidone hydrochloride has been reported: The condensation of 6-chloroindolin-2-one (I) with bromoacetic acid (II) by means of polyphosphoric acid (PPA) gives 5-(bromoacetyl)-6-chloroindolin-2-one (III), which is reduced with triethylsilane and trifluoroacetic acid to the corresponding 2-bromoethyl derivative (IV). Finally, this compound is condensed with 4-(3-benzisothiazolyl)piperazine (V) by means of Na2CO3 in DMF or isobutyl methyl ketone.
……………………….
http://www.google.com/patents/WO2004050655A1?cl=en
REFERENCE EXAMPLE 1 Preparation of 5-(2-Chloro ethyl) -6-chloro oxindole
Triethylsilane (57.2 gm) was added slowly to the reaction mixture of 5-(2-
Chloro acetyl) -6-chloro oxindole (50.0 gm) and trifluoroacetic acid (175 mL) below the temperature of 45°C. The reaction was maintained at 40-45°C for 6 hours. The reaction mass was cooled to 0°C to -5°C and maintained stirring for 90 minutes. The separated solid was filtered and washed with water (50 mL). Then the wet compound was further slurred in water (250 mL) for 90 minutes. The resultant solid was filtered, washed with water (50 mL) and dried at a temperature of 70-75°C to afford 5-(2-chloroethyl) -6-chloro oxindole (43.5 gm).
REFERENCE EXAMPLE 2 Preparation of Ziprasidone base
Refluxed the reaction mixture of 5-(2-chloroethyl) -6-chloro oxindole (100 gm), 3-(l-piperazinyl)-l,2-benzisothiazole (104.7 gm), sodium carbonate (92.2 gm), sodium iodide (6.4 gm), terra butyl ammonium bromide (28 gm) and cyclohexane (1000 mL) till the reaction was completed. The reaction mass was cooled to a temperature of 30°C and the solid was filtered. To the wet compound was added water (1000 mL) and continued stirring for 45 minutes. The solid was filtered and washed with water (100 mL).
To the water wet compound was added acetone (500 mL) and there was stirring for 2 hours at room temperature. The compound was filtered and washed with acetone (200 mL) and dried at a temperature of 70-75°C to afford the Cmde Ziprasidone base (156.9 g) REFERENCE EXAMPLE 3
Preparation of Ziprasidone base
Charged 5-(2-chloroethyl) -6-chloro oxindole (50 gm), 3-(l-piperazinyl)-
1,2-benzisothiazole (47.5 gm) and cyclohexane (500 mL) into an autoclave. To this sodium carbonate (46 gm), sodium iodide (3.2 gm), terra butyl phosphonium bromide (14.8 gm) was added and the reaction was maintained at a temperature of 95-102°C and the pressure was kept at 2.5 kg/cm till the reaction was completed. The reaction mass was cooled to 30°C and water (250 mL) was added. The resulting compound was filtered and washed with water (100 mL). The wet compound was further slurred in water (500 mL), filtered and washed with water (100 mL). To the water wet compound was added acetone (500 mL) and was stirred at room temperature for 2 hours and 30 minutes. The solid was filtered, washed with acetone (100 L) and dried at a temperature of 60-65°C to afford the Ziprasidone base (65.7 gm).
EXAMPLE 1
Preparation of amorphous form of Ziprasidone hydrochloride Ziprasidone (5g) and 50 mL of acetic acid were placed into a round bottom flask and heated to 45 – 50°C. Added was 25 mL of aqueous hydrochloric acid slowly to the mixture over 20 min. Then the reaction mixture was refluxed. Water (10 mL) was added, followed by addition of 50 mL of Isopropanol. The reaction mass was cooled to 50°C and distilled off the solvent completely under vacuum. The material formed was scratched from the flask.
EXAMPLE 2
Preparation of crystalline form of Ziprasidone
Sodium carbonate (56.3 g) and 500 mL of water were placed into a round bottom flask. Added was 50 g of 3-(l-piperazinyl)-l,2-benzisothiazole hydrochloride and 50 g of 6-Chloro-5-(2-Chloroethyl) oxindole. The reaction mixture was then refluxed for
15 hours. The reaction completion was monitored by TLC. The reaction mass was cooled to room temperature. The resulting compound was filtered and washed with 50 mL of water. The wet compound and 250 mL of acetone were placed into a flask and the reaction mixture was sthred at room temperature for 2 hours. The reaction mixture was filtered to give a solid cake, which was washed with 50 mL of acetone. The wet cake and
750 mL of methanol were placed into a flask, which was heated to 50°C, and 14 mL of methane sulfonic acid was added to the solution over 20 minutes. The resulting reaction mass was cooled to room temperature and was subjected to a filtration to give a solid compound, which was washed with methanol. The wet compound and 750 mL of water were placed into a flask, and then pH of the solution was adjusted to pH 9 with caustic lye.
The reaction mixture was then stirred at room temperature for 1 hour and filtered. The filtered compound was washed with water and dried at 70°C to give 65 g of crystalline form Ziprasidone base.
……………………..
http://www.google.com/patents/US7087611
Ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride), I is a potent neuroleptic agent useful in the treatment of psychotic disorders, schizophrenia, and anxiety diseases. It is currently marketed under the proprietary name of Geodon.

Ziprasidone hydrochloride is known to exist in three crystalline forms; namely, the monohydrate, hemihydrate and anhydrous form as disclosed in U.S. Pat. Nos. 4,831,031 and 5,312,925, both of which are herein incorporated by reference. U.S. Pat. No. 5,312,925 states that ziprasidone hydrochloride monohydrate is substantially hygroscopically stable, thus alleviating potential problems due to weight changes of the active pharmaceutical ingredient during the final formulation process. Nevertheless a very low aqueous solubility is observed for this crystalline form.
U.S. Pat. No. 4,831,031 discloses that arylpiperazinyl-ethyl (or butyl)-heterocyclic compounds II may be prepared by reacting piperazines of the formula III with compounds of the formula IV as follows:

The ‘031 patent indicates that this coupling reaction is generally conducted in a polar solvent, such as a lower alcohol, dimethylformamide or methyl isobutyl ketone, and in the presence of a weak base and that, preferably, the reaction is in the further presence of a catalytic amount of sodium iodide, and a neutralizing agent for hydrochloride such as sodium carbonate. At example 16, the ‘031 patent discloses a process in which a solution of ziprasidone free base is taken up in dichloromethane and then reacted with ether saturated with HCl to afford a precipitate which is subsequently filtered, slurried with acetone and filtered again to give ziprasidone hydrochoride hemihydrate.
Additionally, the methods described in the prior art for the preparation of some of these crystalline forms, for instance ziprasidone hydrochloride anhydrate provide inconsistent reproducibility. For example, ziprasidone hydrochloride anhydrate has been prepared by prolonged drying in air at 50° C. of the corresponding monohydrate form, as disclosed in U.S. Pat. No. 5,312,925. However, repeated attempts to prepare the anhydrous form of ziprasidone hydrochloride in our laboratory by using the above mentioned conditions failed to produce the expected anhydrate and instead ziprasidone hydrochloride having variable contents of water, including that corresponding to the hemihydrate form, were obtained. Furthermore, even when more drastic conditions were used, (i.e., higher temperatures, longer drying times, under vacuum) anhydrous product was still not obtained.
EXAMPLE 3
Preparation of 5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1, 3-dihydro-2H-indol-2-one hydrochloride anhydrate.
To a 3-necked flask equipped with mechanical stirrer, thermometer and nitrogen inlet was added 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one free base (5.0 g) and 1-methyl-2-pyrrolidinone (60 mL) under nitrogen and the suspension was warmed up to 35–40° C. to dissolution. The flask was cooled to about 25° C. A 20.5% anhydrous solution of hydrogen chloride in isopropanol (6.45 g) was added and the mixture was stirred at about 25° C. for about 3 h. The product was collected by filtration on a Buchner funnel. The filter cake is rinsed twice with 10 mL of isopropanol at 20–25° C. and the damp cake transferred to a flask equipped with magnetic stirrer and nitrogen inlet. Isopropanol was added (30 mL) and the suspension stirred at 20–25° C. for about 2 h. The product was collected by filtration on a Buchner funnel. The filter cake is rinsed with 3×10 mL of isopropanol at 20–25° C. and transferred to a drying oven and dried in vacuo at 70–75° C. for 43 h. This afforded 4.52 g of anhydrous ziprasidone hydrochloride. The material contained 3.1% and 0.26% of residual NMP and IPA, respectively as determined by NMR.
………………………..
http://www.google.com/patents/WO2000059489A2?cl=en
5. EXAMPLES 5.1. EXAMPLE 1: SYNTHESIS OF ZIPRASIDONE
To a 125 mL round bottom flask equipped with an N2 inlet and condenser are added 0.73 g (3.2 mmol) 5-(2-chloroethyl)-6-chloro-oxindole, 0.70 g (3.2 mmol) N-(l,2- benzisothiazol-3-yl)piperazine, 0.68 g (6.4 mmol) sodium carbonate, 2 mg sodium iodide, and 30 mL methylisobutyl ketone. The reaction is refluxed for 40 hours, cooled, filtered, and evaporated. The residue is chromatographed on silica gel, eluting the by-products with ethyl acetate (1 L) and the product with 4 % methanol in ethyl acetate (1.5 L). The product fractions (Ry = 0.2 in 5 % methanol in ethyl acetate) are evaporated, taken up in methylene chloride, and precipitated by addition of ether saturated with HC1; the solid is filtered and washed with ether, dried, and washed with acetone. The latter is done by slurrying the solid with acetone and filtering. Ziprasidone is obtained as a high melting, non-hygroscopic solid product having an expected melting point of 288°C to 288.5°C.
……………………
Combined serotonin (5HT2) and dopamine (D2) receptor antagonist. Prepn: J. A. Lowe III, A. A. Nagel, EP281309; eidem, US 4831031 (1988, 1989 both to Pfizer). Clinical pharmacology: C. J. Bench et al., Psychopharmacology 112, 308 (1993). HPLC determn in serum: J. S. Janiszewski et al., J. Chromatogr. B 668, 133 (1995). Receptor binding profile: A. W. Schmidt et al., Eur. J. Pharmacol. 425, 197 (2001). Review of pharmacology and clinical experience: G. L. Stimmel et al., Clin. Ther. 24, 21-37 (2002); P. D. Harvey, C. R. Bowie, Expert Opin. Pharmacother. 6, 337-346 (2005).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4831031 * | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| US5312925 | Sep 1, 1992 | May 17, 1994 | Pfizer Inc. | Neuroleptic agents |
| US5338846 | Apr 20, 1993 | Aug 16, 1994 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds with a piperazine salt |
| US5935960 | Feb 7, 1997 | Aug 10, 1999 | Pfizer Inc. | Treatment of schizophrenia |
| US6150366 | May 27, 1999 | Nov 21, 2000 | Pfizer Inc. | Crystalline ziprasidone free base or crystalline ziprasidone hydrochloride particles with specific particle size are useful as antipsychosis agent |
| US20040152711 | Dec 4, 2003 | Aug 5, 2004 | Dr. Reddy’s Laboratories Limited | Crystal structure and amorphous form; psychological disorders |
| CA2166203A1 | Apr 6, 1994 | Jan 5, 1995 | Processes and intermediates for the preparation of 5-[2-(4-(benzoisothiazol-3-yl)-piperazin-1-yl)ethyl]- 6-chloro-1,3-dihydro-indol-2-one | |
| CA2245269A1 | Aug 7, 1998 | Feb 11, 1999 | Pfizer Prod Inc | Solid pharmaceutical dispersions with enhanced bioavailability |
| CA2252898A1 | Apr 10, 1997 | Nov 13, 1997 | Pfizer | Mesylate dihydrate salts of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one(=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO1995000510A1 | Apr 6, 1994 | Jan 5, 1995 | Pfizer | Processes and intermediates for the preparation of 5-[2-(4-(benzoisothiazol-3-yl)-piperazin-1-yl)ethyl]-6-chloro-1,3-dihydro-indol-2-one |
| WO2003070246A1 | Feb 17, 2003 | Aug 28, 2003 | Pfizer Prod Inc | Controlled synthesis of ziprasidone and compositions thereof |
| WO2004050655A1 | Dec 4, 2003 | Jun 17, 2004 | Akundi Surya Prabhakar | Polymorphic forms of ziprasidone and its hydrochloride |
| US7939662 | Dec 6, 2007 | May 10, 2011 | Apotex Pharmachem Inc. | Amorphous ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride) and processes to produce the same |
| US4831031 * | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 * | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| WO1997042190A1 * | Mar 26, 1997 | Nov 13, 1997 | Frank R Busch | Mesylate trihydrate salt of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one (=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO1997042191A1 * | Apr 10, 1997 | Nov 13, 1997 | Pfizer | Mesylate dihydrate salts of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)-ethyl)-6-chloro-1,3-dihydro-2(1h)-indol-2-one (=ziprasidone), its preparation and its use as dopamine d2 antagonist |
| WO2003099198A2 * | May 26, 2003 | Dec 4, 2003 | Srinivasu Kilaru | A process for the preparation of oxindole derivatives |
| WO2004050655A1 * | Dec 4, 2003 | Jun 17, 2004 | Akundi Surya Prabhakar | Polymorphic forms of ziprasidone and its hydrochloride |
| US6110918 * | Mar 26, 1997 | Aug 29, 2000 | Pfizer Inc | Mesylate trihydrate salt of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihy dro-2(1H)-indol-2-one (=ziprasidone), its preparation and its use as dopamine D2 antagonist |
| US6245765 | Apr 10, 1997 | Jun 12, 2001 | Pfizer Inc | Psychological disorders |
| US7087611 | Aug 30, 2004 | Aug 8, 2006 | Apotex Pharmachem Inc. | Reacting ziprasidone with hydrochloric acid in solvent; salt formation; drying |
| US7488729 | Dec 4, 2003 | Feb 10, 2009 | Dr. Reddy’s Laboratories Limited | Polymorphic forms of ziprasidone and its hydrochloride salt and process for preparation thereof |
| US7790886 | Jan 5, 2009 | Sep 7, 2010 | Dr. Reddy’s Laboratories, Inc. | providing alcoholic solvent, desolventizing to form solid mass, isolating amorphous form; psychosis |
| US7939662 | Dec 6, 2007 | May 10, 2011 | Apotex Pharmachem Inc. | Amorphous ziprasidone hydrochloride (5-[2-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]ethyl]-6-chloro-1,3-dihydro-2H-indol-2-one hydrochloride) and processes to produce the same |
| US8097610 | Aug 24, 2006 | Jan 17, 2012 | Shionogi & Co., Ltd. | Derivative having PPAR agonistic activity |
| US4831031 † | Jan 22, 1988 | May 16, 1989 | Pfizer Inc. | Aryl piperazinyl-(C2 or C4) alkylene heterocyclic compounds having neuroleptic activity |
| US5206366 † | Aug 26, 1992 | Apr 27, 1993 | Pfizer Inc. | Process for preparing aryl piperazinyl-heterocyclic compounds |
| US5312925 † | Sep 1, 1992 | May 17, 1994 | Pfizer Inc. | Neuroleptic agents |
| US6111105 † | Oct 11, 1996 | Aug 29, 2000 | Pfizer, Inc. | Chemical intermediates for ziprasidone, a neuroleptic agent; reacting a 2-(piperidino) or (1,2-benzisoxazol-3-yl) or (2-cyanophenyl)thio),3-cyanobenzene with piperazine at a temperature from 80-170 degrees c. |
| US6245765 † | Apr 10, 1997 | Jun 12, 2001 | Pfizer Inc | Psychological disorders |

